
Quantification of the escape from X chromosome inactivation with the million cell-scale human single-cell omics datasets reveals heterogeneity of escape across cell types and tissues
Published in bioRxiv, 2023
Recommended citation: Yoshihiko Tomofuji†, Ryuya Edahiro, Yuya Shirai, Kian Hong Kock, Kyuto Sonehara, Qingbo S. Wang, Shinichi Namba, Jonathan Moody, Yoshinari Ando, Akari Suzuki, Tomohiro Yata, Kotaro Ogawa, Ho Namkoong, Quy Xiao Xuan Lin, Eliora Violain Buyamin, Le Min Tan, Radhika Sonthalia, Kyung Yeon Han, Hiromu Tanaka, Ho Lee, Asian Immune Diversity Atlas Network, Japan COVID-19 Task Force, The BioBank Japan Project, Tatsusada Okuno, Boxiang Liu, Koichi Matsuda, Koichi Fukunaga, Hideki Mochizuki, Woong-Yang Park, Kazuhiko Yamamoto, Chung-Chau Hon, Jay W. Shin, Shyam Prabhakar, Atsushi Kumanogoh, & Yukinori Okada†. Quantification of the escape from X chromosome inactivation with the million cell-scale human single-cell omics datasets reveals heterogeneity of escape across cell types and tissues. bioRxiv 2023.10.14.561800 (2023) doi:10.1101/2023.10.14.561800. https://www.biorxiv.org/content/10.1101/2023.10.14.561800v1

One of the two X chromosomes of females is silenced through X chromosome inactivation (XCI) to compensate for the difference in the dosage between sexes. Among the X-linked genes, several genes escape from XCI, which could contribute to the differential gene expression between the sexes. However, the differences in the escape across cell types and tissues are still poorly characterized because no methods could directly evaluate the escape under a physiological condition at the cell-cluster resolution with versatile technology.
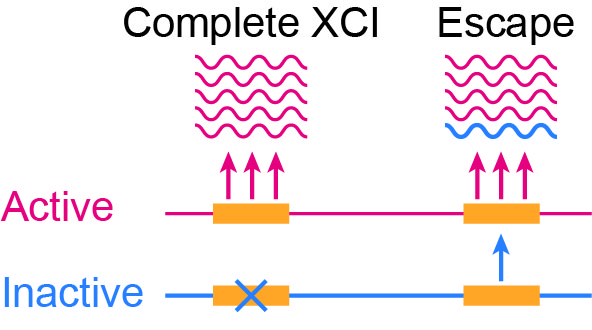
Here, we developed a method, single-cell Level inactivated X chromosome mapping (scLinaX), which directly quantifies relative gene expression from the inactivated X chromosome with droplet-based single-cell RNA-sequencing (scRNA-seq) data.
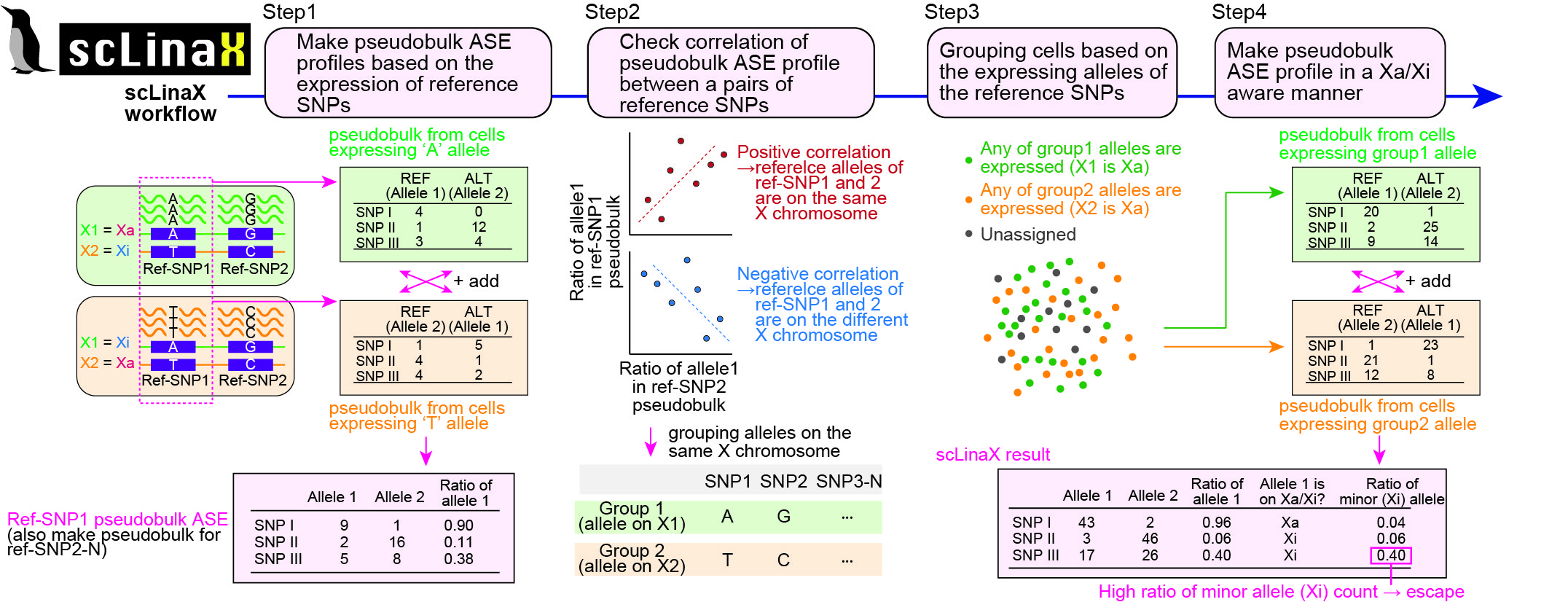
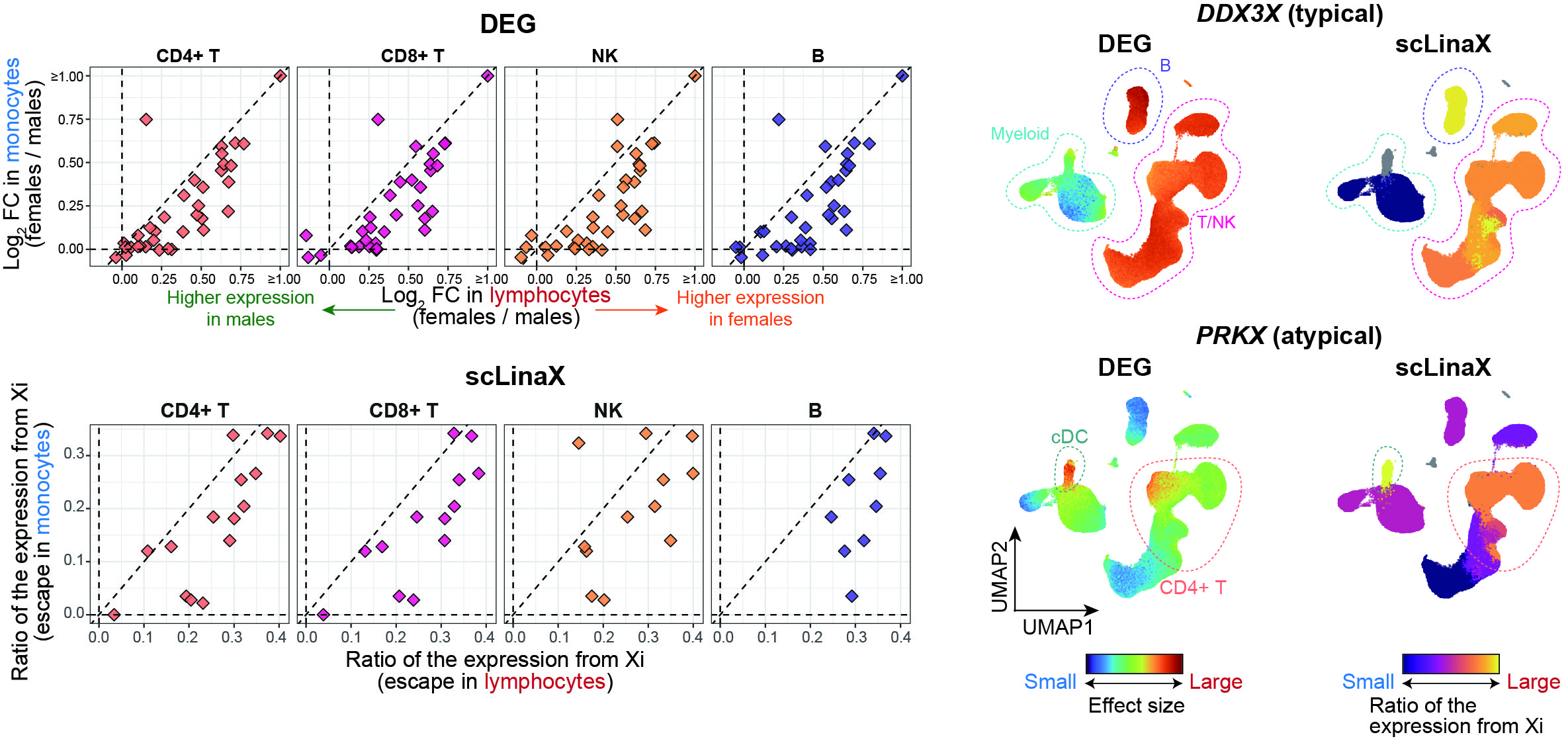
The scLinaX and differentially expressed genes analyses with the scRNA-seq datasets of ~1,000,000 blood cells consistently identified the relatively strong degree of escape in lymphocytes compared to myeloid cells.
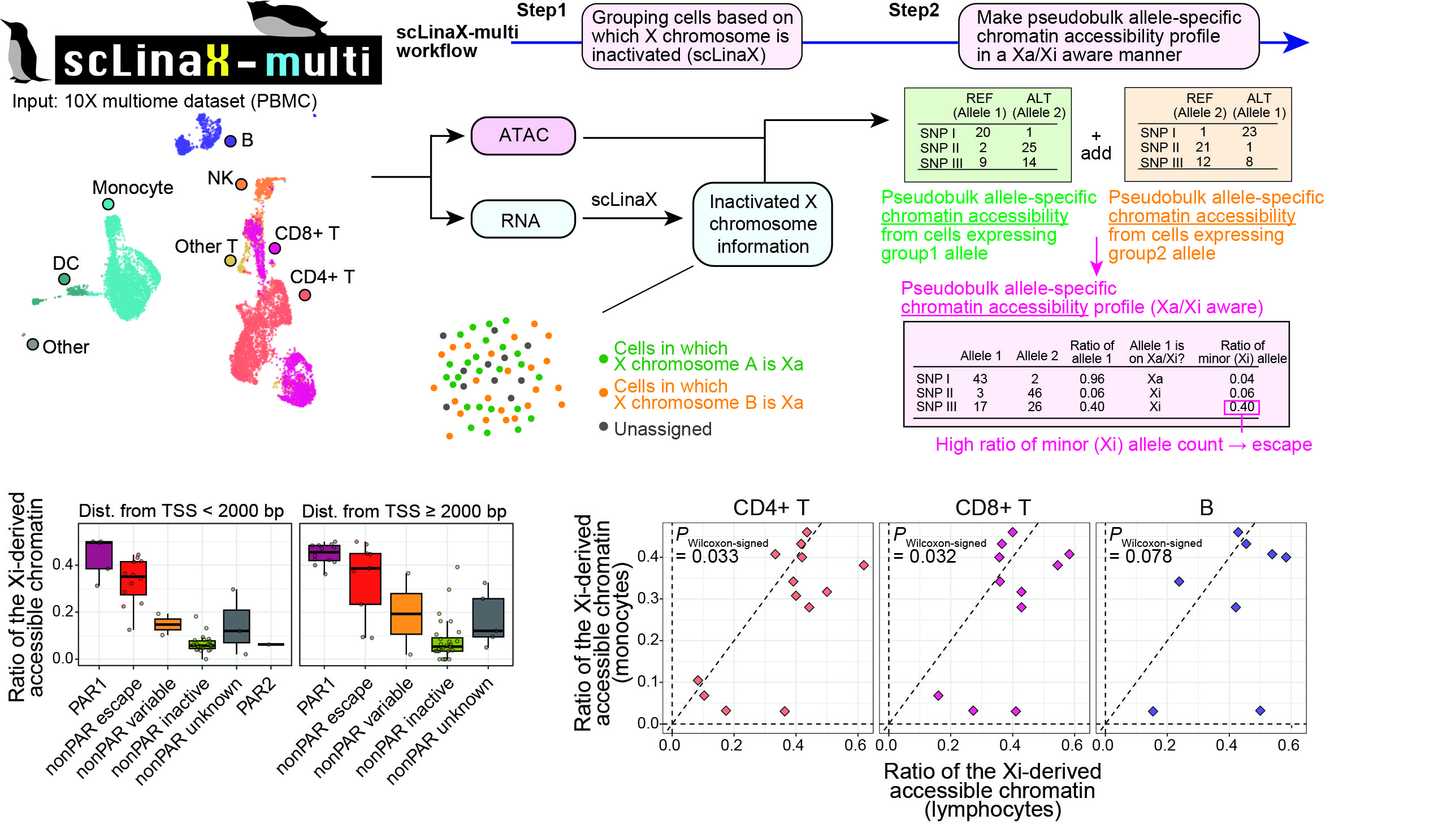
An extension of scLinaX for multi-modal datasets, scLinaX-multi, suggested a stronger degree of escape in lymphocytes than myeloid cells at the chromatin-accessibility level with a 10X multiome dataset.
The scLinaX analysis with the human multiple-organ scRNA-seq datasets also identified the relatively strong degree of escape from XCI in lymphoid tissues and lymphocytes.
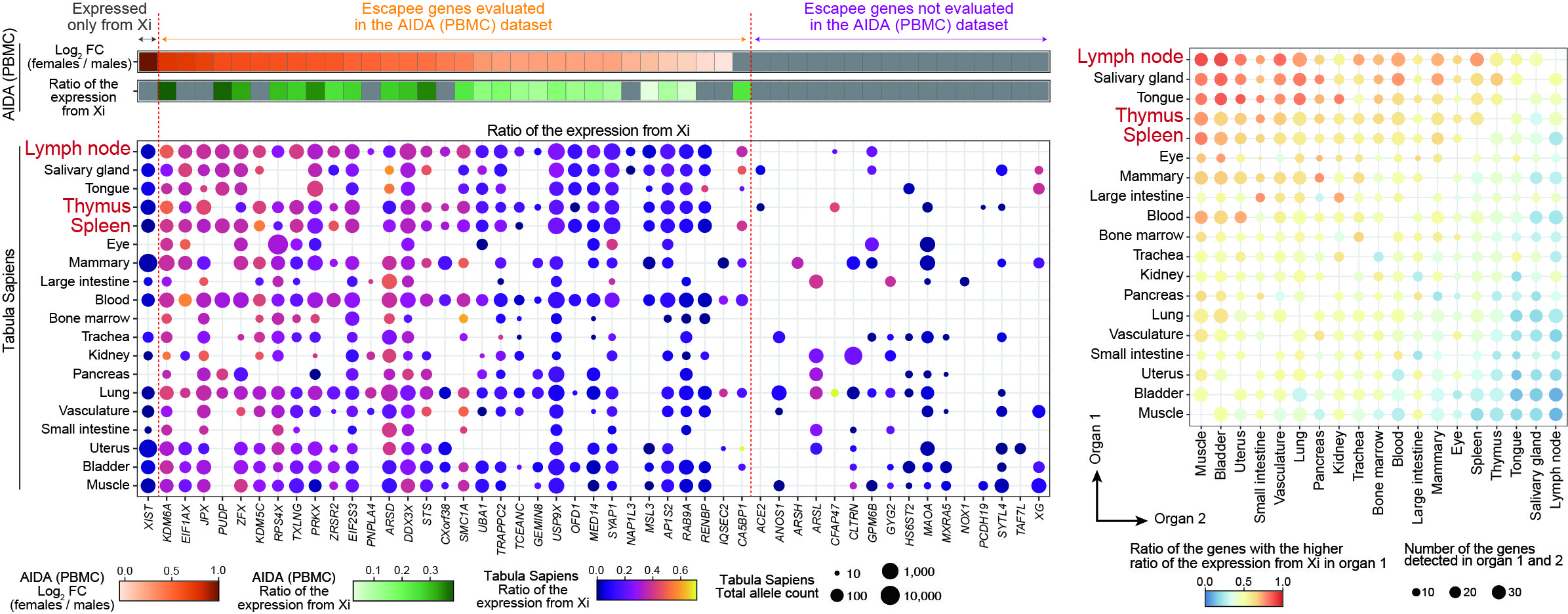
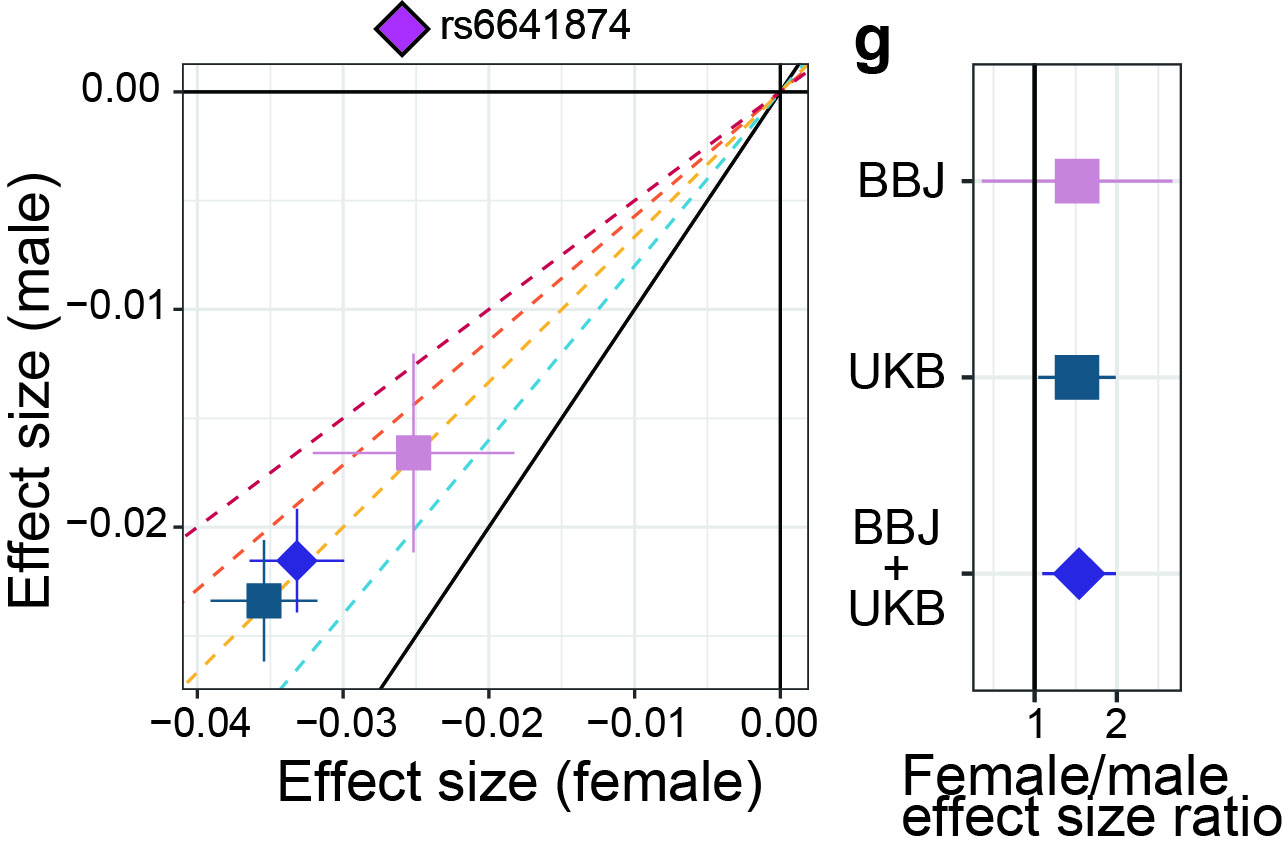
Finally, effect size comparisons of genome-wide association studies between sexes identified the larger effect sizes of the PRKX gene locus-lymphocyte counts association in females than males. This could suggest evidence of the underlying impact of escape on the genotype–phenotype association in humans.
Overall, scLinaX and the quantified catalog of escape identified the heterogeneity of escape across cell types and tissues and would contribute to expanding the current understanding of the XCI, escape, and sex differences in gene regulation. scLinaX R package is available from Github .